This image and one below is taken from the Sequencing.com website. Will Powers got his PFS patients to get Whole Genome Sequencing done through sequencing.com. He read the resulting information using gene.iobio.
The ‘system’ – ‘powers-that-be’ is/are unhappy with developments like this for several reasons. One concern is that our genetic data may reveal risks of future illnesses which, if the data fell into the hands of an insurance company, might lead to an increase in our insurance premiums. Laws and regulations have been put in place to protect us against this.
Another concern is that a ‘cowboy’ reading of specialist data might lead to all sorts of extravagant claims on the back of which people take risks they should not take. Again there are good grounds to worry about this.
All of this makes sense but it comes with a hazard. It also risks handing to others the power to interpret material that has come from us in a manner that may suit system interests rather than ours. At the end of the day, actions need judgement calls – decisions. Data doesn’t decide. For us to decide, we need some familiarity with the ingredients going into a decision so we can decide rather than get forced to follow what someone else says.
Among the most striking features of the recent Enduring Sexual Dysfunction Congress was the extent to which Will Powers’ genetic findings supported what individuals affected by PSSD and PFS said about what happens them – details that no clinical trials or other research have so far supported.
Regulations, however, risk leading to Will’s studies getting closed down. We may need to find other ways to continue what he has begun.
Screening Genes
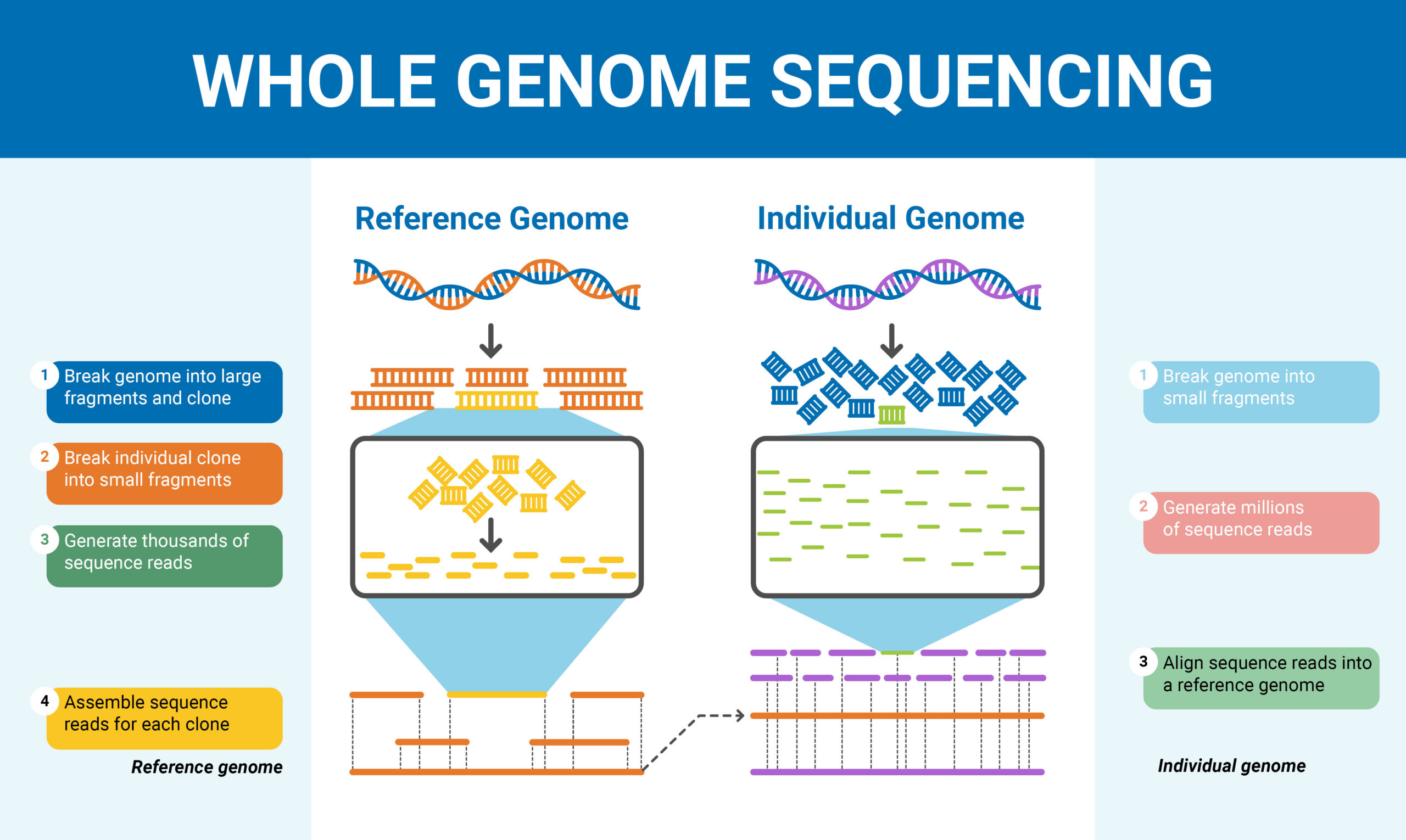
If you are like me, you know little or nothing about genes or about coding computers. But apparently among other things, AI has got to the point of being able to create Apps that can grapple with a lot things previously beyond the reach of ordinary folk like me.
The following post is taken from a set of comments by Peter Grace linked to last weeks Ending Enduring Sexual Dysfunctions post.
Peter Grace
I started thinking of ways to solve the DNA data privacy issue. It occurred to me that if we can’t have people shooting VCF files over gmail without moving operations to Kazakhstan, maybe we could make the data more abstract. Instead of looking at people’s genomes, is there an abstract way to just get a “show of hands” for variant lists of interest without seeing the bodies raising those hands. All that is shared is an integer, a count.
Searching around I found that this protocol already exists its called Beacon: See also https://www.ga4gh.org/product/beacon-api/
It’s used by institutions but I thought perhaps there is no reason we couldn’t use the protocol.
How it might work specifically:
This proposal is for a small, simple system that lets the community ask and answer research questions like that at scale, while keeping every individual’s genome firmly on their own computer.
Someone in the community, I’m imagining it would be a researcher, curates lists of genetic variants worth investigating. They write up a simple text file containing those variants and post it on a suitable public platform, it might be a curated Github page, or any platform that can be curated. The file might be called something like “PSSD Variant list 14”.
Anyone who has their genetic data and wants to participate downloads the list and runs it through a small custom application on their own computer. The app loads their genome file, checks how many of the listed variants are present, and shows them a count: “You have 3 of these 12 variants.” That’s the moment the user decides whether to contribute. If they want to, they submit the number, just the number, “3”, to a public tally for that query. If they don’t, they close the app and nothing happens.
After enough people have submitted, someone tallies the responses and posts the aggregate finding: “Of 47 self-identified PSSD sufferers who ran Query 14, the average count was 1.8, with 31 people having at least one of the listed variants present.”
The assumption would be that self-selection pressure would ensure that mostly people who genuinely suspect they have the phenotype of interest will download the file and submit their count. You could further encourage this by providing clear instructions about the purpose of each list file and the phenotype it relates to on the website where the files are hosted.
No user information is shared, the researcher doesn’t see where the counts came from, they just see the raw tally.
Python has an existing library for parsing Variant Call Format (VCF) files and the application would be quite simple, just a GUI that lets users load up the list file, run the search and submit their count to the server. The only skills users would need would be the ability to download the application and enough of a clue to know which one the VCF file is. I could probably build such an application, not that I’m any great programmer but it doesn’t need to do much.
Beacon V2 has some extra safety rails to prevent accidental leaks: query rate-limiting and minimum-N suppression. Query rate limiting prevents a leak by a Guess Who/Celebrity Heads kind of situation where you leak by posing too many overlapping variant queries. But it might not be needed in our use case as we would be convenience sampling a population of unknown size rather than a fixed institutional database, that is provided the variant query files were hosted on a site accessible to the entire population, not just members of a Reddit or Facebook group. You can’t play Guess Who if you don’t know the number of faces on the board. Minimum-N suppression might still be needed but that is a no-brainer.
Of course it’s just one possible method of abstracting the data for privacy based on Beacon, we might be able to come up with something better. It is also possible an existing platform could be repurposed for this sort of use.
It may be that the moment you upload the VCF file to anything on the internet you then open up all the legal issues, but Gene.iobio is online and they apparently get around part of the legal issue by only streaming the parts of the genome that are being queried at any one moment so the whole genome is never stored “online” at once.
Depending on the institutional involvement we might not even need to be as careful as I am proposing. If a private individual posts a variant list online and another private individual runs the list against their own genome on their own private computer and then posts online that they had matches for this or that variant, has any privacy law been violated?
If its an instituition posting the variant lists and people are replying with their results to a practicing clinical researcher, that I think is more where the problem would be. But I think if you ask around between the tech savvy and legal savvy you’ll be able to find a way of going about it.
Just making it easier for people to run a variant checklist on their own computer might be helpful for people who can use a computer OK but don’t feel confident with tools like gene.iobio. It also prevents any confusion and people coming back saying “I looked and I have all of those genes!”. It might make it easier to roll out for controls as well, which we may still wind up needing.
Vincent Schmitt
Looking at PG’s comments said;
Yes it seems the right idea. Some app running on the desktop/phone of the patient. He would be in control of his data and also the app.
Maybe the code for data extraction/computation from the data should be ”open” so that one can check there is no spying on confidential information.
But on the server side, if you want to collect data for stats, you should check the consistency of data. Imagine one pharma company wanting to spoil your data…
[I can’t see why a pharma company would want to spoil the data – DH – PSSD is a problem that pharma would like to see go away].
Next Steps
I/We (many of us) need people who are much better versed in these things than we are to scrutinize the options laid out here and offer input on what might usefully be done. Useful in the sense of what might empower us. Rather than useful in the sense of keep us safe (disempower us).
Given that all pharmaceutical companies, all regulators, all politicians and all health service companies shout from the rooftops that their number one priority is to keep us safe, the assumption has to be that they really mean keep them safe from us. There is no reason to think academic institutions, other bureaucracies or anyone has empowering us to keep ourselves safe as their number one priority.
I will attempt to run this by some institutions and see what comes back
Linked
This post links to
Calling Isotretinoin and SSRI Problem Solvers
Ending Enduring Sexual Dysfunctions
and on RxISK
Sequencing.com is not without biases. They promote medications , for example, to prevent Alzheimer’s such as N-Saids, Statins and others. When I see misinformation, lack of evidence, I lose interest in the company.
Jo Ann
We are not endorsing Sequencing.com. It came into the frame because that is what Will Powers used. The post is about creating work-arounds the groups already present and not necessarily serving our interests rather than the interests of others
David
From Vincent Schmitt
Thinking about it. I see a technical challenge with that project. If I understand well the data collection/preprocessing should be done on the patients’ desktop with some
app we provide them with. In this case the data should have some standard format
so that the app can process them. The patient will not bother spending much time
entering data. So is there already some kind of standard format? Getting some fine big data for statistical treatment is often a pain.
This is an excellent point.
Before I get into the technical side I’ll make a point that for participants, the simplest way to think about any application is like a car. The person driving it does not climb into the engine bay. For them, it is a black box: it just does what it says on the tin.
But this question raises a problem for the mechanics and the car manufacturer ie. the researchers.
I would think the best format for the variants of interest list/s would also be VCF, just like the file it would be queried against. But that is the issue highlighted here. I galloped over the part where the researcher needs to be able to generate such a file in the first place. It is not just a matter of using a rsID to refer to the variants; you need the other data required to match things correctly, including which reference genome is being used.
Gene.iobio appears to let users export their variants list as a VCF. That is one part of the workflow solved but its not the final step. A VCF exported from one single patient’s genome, even if it only contains a handful of bookmarked variants, could still disclose something about that patient. Next the researcher would need to take all of the different VCFs they’ve exported and create a cleaned and curated VCF list drawn from a sample of patients. I think that should be enough to avoid the privacy issue, but it would be worth double checking.
So the hard part really is putting together the curated list/s. The researcher/s putting the list/s together would have to bring all the files they have exported from gene.iobio into a spreadsheet and basically cut and paste until they are happy with their final list. I mean, that is an unavoidable task. It is a tedious editorial process. But it is a process that would have to be undertaken anyway regardless of the existence of this app we are discussing. I’d imagine there is a very good chance William already has a couple of messy spreadsheets he is running to track these variants.
It occurs to me that you might have a situation where the variants of interest list/s are very large. I assume if William is tracking things manually he must have a manageable number of candidates. Still, that number could grow, and I don’t have a feel for how big the list/s could get. This is still unavoidable, but it goes towards feasibility and the size of the team needed on the research end. Hopefully it’s manageable for a very small team.
Actually as I’m writing this I see gene.iobio has an option to export lists a CSV files, not sure if that is a CSV with the same headers as a VCF but regardless of the exact best format, the bottom line is someone will have to spend some time putting together those final lists.
Here is an example of the headers that are sent out for a Beacon 2 query.
Request Parameters:
assemblyId
referenceName
start
end variation.location.interval.end
referenceBases
alternateBases
variantType
variantMinLength
variantMaxLength
mateName
gene
aachange
So that is the information that is sent for each variant when a researcher wants to query other databases in the beacon network. We’re doing something similar and the headers of a VCF file are similar to this. So hopefully those headers give you an idea of what our list file would look like opened up as a spreadsheet.
Look how difficult it was for Joanna Le Noury to investigate 77,000 pages of documents which GSK made difficult –
https://study329.org/the-data/
In those days none of the information was ‘encrypted’ – it was just a massive task – today’s genome testing outfits seem to favour encryption, think WhatsApp, to avoid unscrupulous nosying of data.
GSK didn’t reckon on the clever folk with fortitude, getting their machinations in to gear…
It just occurred to me that I should have posted this as a comment – since it’s primarily a response to Peter G and Vincent.
My No 2 son, Jamie, is an algorithm engineer in a complex, high risk industry – not a web builder. But he gets this stuff – it’s a particular way of thinking. I asked him to have a quick read through the blog and comment – from a technical pov – in terms even I could understand. This is his feedback. He’s happy to talk through any issues, is easily tempted to write ‘a bit of code’ etc.
Overview
‘Looks perfectly feasible like this – I’m not a web developer but..
I looked at the (Beacon) protocol and the gist of running it yourself is it takes your files turns them into a different format, it’s called JSON and it puts them into a little database. It’s just a specific format that makes it queryable and then chucks it into a database. That’s already baked in. And then what happens is that database gets queried. That’s the protocol, that’s a little transformation. That’s all established.
It’s set up specifically so that the person who actually holds the data doesn’t really need do a huge amount, doesn’t need to know anything about it. All it does is takes all these files – you only have one file so it’ll be a small database – coverts it, stores it in the database and then it can query the database through this protocol.
Pitfalls
There’s nothing there that seems like it would be hard-you just have to be aware of the pitfalls.
That protocol he’s (Peter) talking about is open source. You just have to be wary about what the license says. There are certain conditions, like some open source means if you use it you have to make your own stuff open source. Sometimes you can use it personally but not for x y and z. I don’t know what their license is- if the protocol’s all open source.
Just be aware. As a user you can download an application for free and use it for free but it doesn’t mean you can modify it in a closed way.
Anonymity etc.
You’ve got your app locally and now you want to send information. What you don’t want is someone sending the same thing multiple times. What you’re able to do is anonymously identify someone. This is NOT how you do it- this is a simple version- say you give everyone a random number, they give you the random number when they give you their file. You do a thing called a hash, which is basically a big random string of stuff which is specifically created from someone’s numbers so that you can’t work out what that thing is or who that person is.
Someone trying to exploit it, or anonymity problems so – what you do is you need to deal with that in a certain way – if you’re just doing a tally as well and not holding any other information – once you take in your bit you can just delete it – you don’t need to link that number to that person. You add that to your tally – and your front end goes we’ve already received the information on this for this specific file.
The bit you do on your computer will be fairly easy, you just have to make sure if you want anonymity how you’re feeding the information back and then doing checks that you are, at some point you are handling it undeleted – trust me this will be deleted – a lot of people say that, but they don’t – you need to be very aware we are doing this.
Maybe everyone that downloads it generates a random number that you use as hashing that identifies you. You might do that and you might also hash it with the file that they’re giving you so you can identify a specific user having used the file so if you receive another one it would be the same thing. Specific for each file they’re doing it on. You generate the hash based on your number and some information on the file. The hash will be different for every single file.
There will be security protocols dealing with all this and a load of stuff already available.
Building the beast – (although to me, H, it sounds more like assembly from what’s already available with a bit of extra code).
You could probably build it in Python – it’s easy and you don’t need the speed and things like that. It kind of depends on what that API is already written in. It lets you interface with a system-send it little messages. Depends what the codes are already in.
The key thing with all of this is to do it step by step. You need an app that implements what you need to do- takes in a file and outputs.
If you implement – how to anonymise, how to interface with the user – that’s just the back end code it will literally be like you’re doing a command line. The general code of the app.
That layer goes around the original.
You need to write a bit of code that does the thing you want to do first then you wrap that code in a wrapper which deals with the user, deals with anonymising and sends it.
You then need to deal how you’re going to receive it. Not my world server world but at that point you’re taking in data in a fixed format and from users or whatever.
H
Thanks for this – I’ll have to leave it for others to wade in and see if a workable copy can be made.
Nest week I should be back in contact with 2 university departments and I may begin to get a sense of what ehey can do for us and also what they make of this people power approach
David
Yes, this is all correct and has 0ne or two details I hadn’t given enough thought to.
Beacon does indeed use JSON but at the moment I’m thinking that the conversion to JSON would happen in the application itself hidden from view. Users would just load in a mini VCF file, it would get converted to JSON and then parsed into a query. But now I’m getting into the weeds.
He raises an important issue around open source. I imagine that open source would be the best way to do this because allowing everyone to see how the application works is really the only way I can think of to provide users with the kind of proper informed consent to keep everything above board and more importantly, the application will itself be accessing their genome data, so people would need to be able to look at the source code to make sure no one has added a bit of extra code that does something nefarious. But yes, we might need to give some consideration to the exact open source license.
The other point that I thought of after I wrote the idea is that of course the application would need to connect to the internet to send the beacon back. Now, that is another reason to make it open source and transparent but it also means we would need some way to authenticate the users without identifying them. A digital handshake, a random serial token generated for each exchange. It would also need careful data validation.
One small practical addition might be to collect structured exposure/phenotype metadata through a short questionnaire alongside the variant count: suspected triggering drug, other relevant exposures, approximate medication order, time to symptom onset, main symptom domains, and whether the history seems to involve one main exposure or several.
This could help interpretation later, especially when histories involve successive exposures — for example isotretinoin followed by finasteride after hair loss, and/or antidepressants after depression, anxiety, or during subsequent consultations.
A related privacy point might be worth keeping in mind: patients may also need clarity at the sequencing-provider level — where raw data are stored, whether they can be deleted or reused, and what happens if the company is acquired, breached or goes bankrupt, as the 23andMe case reminded many people.
Taken together, local VCF analysis, open-source validation, upstream privacy clarity and structured exposure/phenotype metadata could help make the approach safer and easier to interpret.
While keeping an account of exposures may seem to make sense, it may only do so in the case of isotretinoin and it is unlikely that many if any isotreatinoin patients are going to get involved in this research.
Very very few people with PSSD have had exposure to isotretinoin or finasteride. Some with PFS may subsequently get an SSRI – RxISK has a very small number of cases – but they view themselves and are reasonably viewed as PFS cases whose genetic variants prior to SSRI used caused the problem.
D
What I really need to get my head around is William’s actual workflow in Gene.ioboi. I need to understand what kind of dataset we’re dealing with, get a feel for its size. If its big and unwieldy this app makes sense but if he’s talking about a truly small number of variants maybe we don’t even need an app.
The main advantage of something like an application would be to eliminate false positives and other extraneous variables, which are a major issue with informal sharing of variant information. Just a few weeks ago I saw an example where someone had run a report through AI and it told them they had the risk alleles for bipolar. In reality the report simply mentioned the genes and closer inspection revealed the variants listed for that genes were the low risk ones. So sending people off to look for themselves is fraught but there may be other research methods, manual systems that might work if well organised and if the dataset is small enough. Toyota still run their whole factory on kanban, so not everything has to be done on a computer to succeed, it just needs to be operationalised.
I had a response from Will that says:
This is not genetic genie, that’s a different website.
Gene.iobio is where I load up a 300gb file that contains literally every codon in a person’s genome start to finish. I then go through the relevant genes manually. very rarely is it an obvious stop codon or something like that. I am personally looking at each variant and making an educated decision about whether or not it matters based on what amino acid swap it is, the revel score, where in the protein it is, and the rarity of the mutation.
This requires actual thought, and as of right now, there isn’t an AI to do this.
I could easily have an AI scan these genomes for the super well known “most common” style mutations, or obvious gene deletions. But deciding whether or not a 0.55 revel score mutation in X part of Y gene with an incidence rate of 1/5000 people matters is still a human’s job at this time.
looking at the genomes seems exciting. If it logistically works out, what would happen next? Who would analize the data. I understand dr Powers is very busy. Are there getting more gene “wizards” involved?
We’re trying to find some others
D