
This post has a partner – There was a Young Woman who Swallowed a Lie. It also needs to be read with its Health Warning.
A Full Eclipse of Care
In the early 1980s, the pharmaceutical industry outsourced the running of its randomized clinical trials (RCTs) to Contract Research Organizations (CROs) and the writing up of RCT results to medical writing companies.
With this, the physicians and academics running trials of drugs and vaccines lost access to clinical trial data. Even the apparent authors on the manuscripts representing the results of those trials no longer had access to and could not interrogate the data or stand as guarantors for the accuracy of the published record of these trials.
Study 329, a study comparing paroxetine to placebo in depressed minors conducted in the mid-1990s, illustrates the changes then taking place. A 2001 article with a distinguished authorship line, in the journal with the highest impact factor in child psychiatry, claimed paroxetine worked well and was safe. It led to a considerable increase in the prescribing of paroxetine to minors.
Based on documents I provided, in 2004 New York State took a fraud action against the makers of paroxetine, GlaxoSmithKline (GSK), which was resolved with a promise to make company trial data more available. This high-profile action led to Black Box Warnings on antidepressants. It made clear that the entire published literature on RCTs of antidepressants in children at that time was ghostwritten – the names on the authorship lines for medical paper reporting the results of RCTs do not write those papers. It also laid bare a comprehensive mismatch between the published claims and the data when accessed.
Study 329 and paroxetine were also the centrepiece of an action the US Dept of Justice took against GSK that was resolved in 2012 with a payment of $3 billion then the largest such payment in corporate history.
Study 329 was not an aberration. It represents standard industry modus operandi as of the mid-1990s. It was performed in the best academic centres with good oversight, and to higher standards than most trials are now.
These legal actions made Study 329 trial data uniquely available. This permitted colleagues and I some years later, to Restore Study 329 and demonstrate the range of tricks companies use in hiding harms.
Most of the tricks companies now deploy to hide harms and fudge efficacy were being worked out at the time this trial took place. Company abilities to hoodwink us have improved greatly since then and there is likely more fraud now than then.
A recent BMJ publication on Pfizer’s vaccine trials illustrates that these trials are affected by the practices that were becoming standard in the 1990s.
Besides company tricks, even if done by angels, as pointed out by Austin Bradford Hill who ran the first randomized trial, RCTs can be helpful in evaluating one of the more than one hundred things every drug does but this, by definition, makes them a poor way to evaluate a drug or vaccine overall.
The need to supplement RCTs with other evaluative approaches can be brought out by a simple example. Before the selective serotonin reuptake inhibiting (SSRI) group of antidepressants were put into clinical trials, it was known these drugs affect sexual functioning in close to 100% of us within 30 minutes of a first pill. But in RCTs of these drugs undertaken for the purposes of establishing the existence of a much less common benefit for nervous problems, the focus on a primary endpoint that RCTs require meant these sexual effects essentially vanished.
The vanishing-effects problem is even greater in current vaccine trials, where participants have been presented with a prepopulated list of a small number of side effects that might happen to them as a result of the vaccine (see New England Journal of Misinformation and appendices 2 and 3). This is like a company claiming an improvement in its consumer satisfaction levels after eliminating the complaints department.

Consent
Two issues arise when treatment effects, other than those of commercial interest, vanish. Because of a premium now put on RCTs, companies claim that the only things that happen on a drug or vaccine are things that happen to a statistically significant effect in an RCT. Everything else is anecdotal or psychogenic.
Statistical significance testing, however, should only be applied to the primary endpoint of a trial. It should not be applied to effects that are not being investigated.
As a result, the consent forms for people taking Covid vaccines, in the UK for instance, now present people with a strong steer that the only things likely to happen them after injection are a sore arm, headaches or other aches, fatigue, stomach upset, or a mild fever.
Pfizer knew about the effects of SSRIs on sexual functioning and risk of triggering suicidality before launching sertraline. The company similarly knew about the risks of its vaccine to individuals with pre-existing neurological conditions such as multiple sclerosis, or the more general risk of effects on the nervous system such as Guillain-Barre Syndrome, Bell’s Palsy, demyelinating disorders or transverse myelitis, all of which people should be warned about prior to deciding whether to take the vaccine or not.
Risk-Benefit
A second issue stems from the current misreading of what RCT data demonstrate, namely that it is not possible on the basis of RCTs to establish a Risk-Benefit ratio for a drug or a vaccine.
Regulators routinely claim that the Risk-Benefit ratio favours the approval of this drug or vaccine or not warning about harms, basing this claim on company RCT data and assuming the primary endpoint is by far the commonest effect of treatment with all other effects being rare or idiosyncratic or developing outside the time frame of the trial.
But if RCTs can make effects of a drug or vaccine that are as common or even more common than the primary endpoint vanish, or if other effects are rare or idiosyncratic but serious, then the basis for claiming a favourable Risk-Benefit ratio for drugs or vaccines vanishes.
This clearly is likely to be even more the case for novel mRNA technologies.
There is furthermore no metric, or algorithm, for making Risk-Benefit judgements on a population level. These are a matter of individual decision.
Pharmacovigilance
The development of modern therapeutics has put pharmacovigilance at the heart of medicine. While there are epidemiological and other processes that pharmacovigilance can turn to, the central discipline involves a doctor and patient deciding on the basis of an examination of the patient what is happening when the patient reports some change after taking a medicine. The event being considered could be a benefit in which case doctor and patient will opt to continue treatment. Unless such decisions are ordinarily correct, medical practice could not continue.
Or the event could be that the treatment is not working or is causing a harm.
This assessment by doctor and patient is or should be a judicial process. Judicial processes are commonly viewed as something distinct from science. A judicial process, however, necessarily adheres to available data, and only available data, just as closely as science does. Judicial processes have rules of evidence as strict as science. Speculation has no part in a judicial process. Nor since the execution of Walter Raleigh in 1618 does hearsay. If a witness cannot be brought into the examination room and cross-examined, their evidence is discounted.
The explanation, the verdict, must match the facts presented. It must also achieve a consensus that overcomes the biases of 12 different jurors.
In a clinical encounter, just as in a legal process, the answer is provisional, as are all answers in science. Further facts may come to light that challenge a provisionally accepted view.
As outlined here, this is a process that is as scientific as any of the demonstrations of physical or chemical phenomena undertaken in the Royal Society from 1660 onwards that we view as establishing the scientific paradigm, namely that science seeks to explain observable data, that it challenges bias by sticking to this rule and that all its verdicts are intentionally provisional with the process encouraging others to experiment further.
Good clinical practice, when undertaken as outlined here, is inherently scientific. More scientific than any practice shaped by unavailable RCT data, misleadingly represented in ghostwritten publications that hype the benefits of a treatment and hide the hazards.
Clinicians, who attempt to be scientific in this manner, however, are increasingly called on to account for the mismatch between their judgements based on what they see in and hear from patients, for instance that this SSRI made this person suicidal, and the denial of this possibility in an apparently scientific literature, along with the NICE or other guidelines that are based on this literature.
Regulation and Pharmacovigilance
The goal of medicines regulation, as in traffic, financial, or airline regulation, is to enhance safety. It is not to enhance efficacy.
A set of Amendments to the US Food and Drugs Act put in place in 1962 introduced RCTs to the regulation of medicines for the contribution they might make to assessing therapeutic efficacy, in the hope that eliminating ineffective treatments might contribute to safety.
Safety, however, has not been enhanced by these regulations, at least not since the 1980s. Prior to 1962, the benefits of treatment had to be evident to clinicians and patients, whereas now, if a sufficiently large number of patients are recruited to a trial, statistically significant effects can be established on surrogate outcomes with these leading to approvals without any doctors or patients seeing an evident benefit, and with more deaths on treatment than if the patient were left untreated.
It was thought in 1962 that clinicians would remain, as they had been before, the principal agents in medicines’ regulation, with the bureaucrats we now call regulators continuing to occupy a relatively minor role.
In contrast to clinicians, the bureaucrats working in the United States Food and Drug Administration (FDA) and Britain’s Medicines and Healthcare Regulatory Agency (MHRA) do not engage with the judicial processes outlined above for four reasons.
1). When doctors or patients report adverse events to MHRA, the first step, if the reporting process has not already achieved this, is for the bureaucrat to strip the names of patients from any communication – ostensibly to comply with clinical confidentiality requirements. This transforms reports from doctors (or patients) into Hearsay.
Anonymization makes reports from injured patients and their doctors inadmissible as evidence in court in a way that case reports with a doctor and patient’s name on them remain admissible. A patient who has been injured by treatment, and further injured by anonymization, cannot be examined and cross-examined and it is not possible to establish cause and effect.
In the case of the Covid vaccines, MHRA, despite huffing and puffing about searching night and day for the needles of causation in a haystack of reports, are faced with a needlestack of reports but are unable to determine any cause-and-effect relationships. While regulators have conceded a link between current vaccines and both thromboses and myocarditis, this was only after clinicians established that these are happening based on their assessment of patients in front of them. To save face regulators have had to agree.
In the event of reports to them, pharmaceutical companies, in contrast to regulators, are legally obliged to follow up patients and their doctors over time to establish whether the company’s drug might be causing a problem. Companies do this and decide their drug has caused a harm and add this harm to their drugs label, while regulators stack up thousands of reports and claim they have never made a causal link in a single case.
2). A further factor inhibits regulators from making causal determinations in respect of deaths on vaccines and drugs. In the case of deaths, regulators like MHRA in the UK ordinarily wait for inquests, before coming to a view. These inquests have inputs from the patient’s doctor. There is also a coroner in place who comes to a view as to what the cause of death has been.
While coroners can indicate a street drug has been a cause of death, they do not have an option (a box to tick) to indicate that a prescription drug or vaccine has caused a death.
Physicians, meanwhile, if asked to attend an inquest or prepare a report on a death, will be advised by their medical insurer to deny a link to any treatment to which they were party. Doctors at inquests are routinely advised in this manner, by the representatives of a business, whose interests lie in not incurring further costs. Containing a medical insurer’s costs is done by deflecting attention from a treatment to an illness.
Coroners, who are normally not medically qualified, are not in a position to gainsay a treating physician who denies a link to treatment. If a coroner is very concerned about a problem, s/he can make a regulation 28 report to MHRA, who will ordinarily opt not to gainsay the view of the treating doctor.
Even the press reporting on inquests are caught in this web, as advice to journalists in the UK, for instance, from the Independent Press Standards Organization (IPSO) on reporting suicides makes clear. IPSO explicitly tells journalists not to go against the conclusion of the coroner, which for the reasons outlined will never implicate a treatment.
3). Regulators support the mantra that RCTs provide gold standard evidence on drugs, partly because RCTs make the bureaucratic job easier. This faith in RCTs extends to claiming that if an effect has not been shown to happen to a statistically significant extent in an RCT, then no matter how plausible it might be, we simply do not know that it happens.
Ian Hudson, the recent CEO of MHRA, said exactly this under oath in the 2001 Tobin v SmithKline trial, when he was working for GSK. The jury rejected the argument.
4). In line with Hudson’s approach, when considering adverse events, FDA and other regulators claim to have dedicated epidemiologists for this task. Epidemiologists have very little role in pharmacovigilance other than in monitoring the risk of birth defects on treatment.
Neither FDA, nor MHRA, have any clinicians trained in assessing adverse event causality. They lack procedures to assess causality. If were they to establish a link between treatment and an event, pharmaceutical companies effectively maintain a drugs label and have to agree before anything happens. Finally, there is reluctance to acknowledge a Suspected Unexpected Serious Adverse Reaction (SUSAR) because of all the work this leads to.
Hearsay
The anonymized reports of deaths and injuries on vaccines are worthless from an evidential point of view in legal and scientific settings, although some value can be extracted from proportional reporting rates.
In contrast, for sixty years the results of RCTs have been taken in legal settings as meeting a Hearsay exemption.
For the first thirty of those sixty years, however, the courts could request the presence of physician authors on the papers reporting the result of company trials, where authorship meant physicians who treated patients who existed with the physician in a position to evaluate the full effect of these drugs in patients.
For the thirty years from 1990 to now, few of the authors on papers reporting company RCTs will have met any of the patients in trials and several authors have ended up in jail for recruiting non-existent patients to trials. Between non-existent patients, and lack of access to the patients who do exist, the results of RCTs arguably should no longer qualify for a Hearsay Exemption.
Regulation and Trial Data
As per scientific norms, there is an assumption that regulators, investigators, and the authors whose names appear on publications of RCT results, see the clinical trial data. None do.
Instead, the regulation of pharmaceutical company products is a business process where commercial confidentiality counts for more than the norms of science. Unlike drugs, company involvement in the case of vaccines is relatively recent. Vaccines were made by countries, who were not constrained by profit considerations.
Companies hold the trial data and submit Clinical Study Reports (CSRs) to the regulator. These are the company representation of what its clinical trial has shown. The CSR will often contain large amounts of largely irrelevant figures set out in Tables. The Tables are in principle transcribed from Clinical Report Forms (CRFs), which, until a recent turn to electronic capture of figures, might have up to 100,000 pages for a 300–400-person trial.
FDA insist they get everything from companies, but if they do get CRFs FDA do not read them other than for audit purposes – checking to see if there are any hints that every twelfth patient might not exist.
As I can attest, based on a more detailed examination of CRFs than FDA undertake, if a significant question mark arises about some harms, FDA will invite the company to revisit its records and tell the regulator what the score is in the light of some concern that has arisen. When matters get this serious, companies are still liable to mislead regulators.
(FDA is mentioned here rather than other regulators because of their claims to thoroughness rather than because they offer a gold-standard in regulation).
More recently FDA led the way in getting what they refer to as individual patient level data (IPD). IPD gives the impression a regulator is getting to grips with something like the raw data but in fact IPD is essentially a spreadsheet with figures primarily linked to efficacy that allow FDA to check on some of the claims for benefits, based on selected data provided. It does not allow FDA or any other regulator to explore hazards or interrogate subjects from trials.
The CSR does include some individual patient level data in the form of narrative reports on serious adverse events (SAE) – events that result in hospitalization or death.
From these narratives, it might be possible to guess that the man whose death was coded as death by burns died because he poured petrol over himself intending to kill himself but only died 5 days later and question the company as to whether this death occurring on a new psychotropic drug rather than placebo might have been better coded as suicide.
But companies have found ways around the obligation to write narratives for SAEs. Patients who drop out of trials because of an adverse event can be designated as having intercurrent illness or some related term. There is no obligation to write a narrative on a patient like this, which leaves anyone who might get to see the records none the wiser as to what has gone on.
In Study 329, a 15-year-old boy was picked up by the police on the street waving a gun and threatening to kill people. He was brought to hospital. This was almost certainly a paroxetine related adverse event. It vanished under an intercurrent illness coding – as did events that befell three other children in this trial, all of whom were on paroxetine and none on placebo.
Ultimately people are the data in clinical trials. Without access to their names and contact details, which should be possible in a vaccine trial where volunteers are not being treated for an illness, no-one can establish what has happened in a clinical trial.
It takes a clinical interview to establish if the person in the trial likely became suicidal as a result of their illness or their drug. The clinical interview is the only place where all the data is present – the later trial database loses data as a result of a semi-automatic allocation of features that an illness and a treatment might share in common, such as suicidality, to the illness.
Up to half of the symptoms that people have in a trial may be features shared in common between a treatment and an illness. This can be teased out in healthy volunteer trials of drugs which remove the illness confounder. In vaccine trials, everyone is a healthy volunteer but companies have found ways to discount the injuries that have happened.
The Eclipse of Medical Science Summary
- Clinicians do not have access to clinical trial data on medicines or vaccines.
- Close to all of the medical literature reporting trial results for on-patent drugs and vaccines is ghostwritten, hyping the benefits and hiding the harms.
- Clinical trials of these treatments that are negative on their primary or their most common outcomes are often published in prestigious journals as positive.
- Clinical trials have their harms airbrushed out of ghostwritten publications.
- Regulators (FDA, Health Canada, MHRA, EMA) do not get to see the full trial data.
- Regulators approve treatments as working even when more people die on active treatment than on placebo.
- Regulators approve medicines on the basis of negative studies and agree not to let the wider world know about this.
- Regulators say nothing when companies publish negative studies as positive and make adverse effects of treatment, including death, vanish.
- For many trials there are more deaths on active treatment than on placebo, but this does not lead regulators to warn about hazards as to do so would in their stated view deter people from seeking a benefit (even when the benefit is better characterized as a commercial benefit to a company rather than a benefit to the individual in terms of a live saved or a restoration of function).
- Regulators do not have pharmacovigilance expertise and a variety of factors inhibit them from linking a treatment to a hazard after that treatment comes on the market.
Pandemic of Overtreatment
Before Covid, there was growing evidence that life expectancies were falling, or improvements in life expectancy had stalled, in many Western countries, including the UK and US. While poverty and inequality kill, as outlined in Shipwreck, or El Naufragio de lo Singular, we also know that a polypharmacy, made possible by the business and regulatory processes outlined above, also kills.
Few of us were on more than a brief course of one medicine per day in the 1980s. Now heading toward 50% of us over the age of 45 are on 3 medicines per day and approaching 50% of us over 65 are on 5 medicines or more per day. The evidence that reducing medication burdens can increase life expectancy, reduce hospitalizations and improve quality of life is strong enough for Health Departments to support efforts in this area.
An unwillingness to tackle the issues raised here, however, semi-mandates a continuing increase in medication burdens, regardless of efforts to reduce meds even in the hopes of saving money. These points are ones that those in favour of vaccine mandates might consider more carefully.
Points 1-10 above have been made to regulators, Ministers of Health, Chief Medical Officers, guideline makers, the editors of medical journals and others. Much of the correspondence is available Here
They have been presented at academic meetings on all continents, featured in articles in major journals and in University Press books without being contested or leading to legal action.
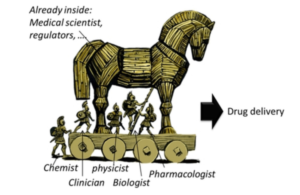
But Health Systems have a bias toward pulling the Horse inside the Citadel.
A Golden Oldie…
GSK’s Transparency and Access Journey
November, 4, 2013 | 10 Comments
https://davidhealy.org/gsks-transparency-and-access-journey/
“At GSK we firmly believe that making more information available, including clinical study reports and anonymised patient-level data, will enable researchers to study the science behind today’s medicines more closely, to learn more about them and how they can best be used.
For GSK the transparency and access journey [i] started with online clinical trial studies in 2004 and has so far led us to the endorsement of the AllTrials Campaign last year, as well as access to anonymised patient data launched in May 2013. Ultimately this has the potential to improve patient care and drive open innovation.
When patients volunteer to take part in research they have a legitimate expectation that the data will be used to enhance knowledge and improve patient care. Our actions towards transparency are an essential part of meeting those expectations. By being open and accessible we become an even stronger and more trusted partner for society, patients and health care professionals and at the same time improve patient care.
We have been open in describing the steps we have taken. Our initiatives has been acknowledged and welcomed by governments, NGO’s and research institutions.
The recently published report from the Science & Technology Committee in UK, states regarding patient level data: “We recognise the efforts of some members of the pharmaceutical industry, particularly GSK, to increase clinical trial transparency and hope that other companies will act in the same spirit in implementing industry-wide principles for responsible clinical trial data sharing”.[ii]
Our initiatives have been commended by campaign groups as setting a new standard for greater transparency. In response to our commitment to develop a system for access to patient level data, Ben Goldacre, (author of “Bad Pharma” and co-founder of AllTrials) said: “It is a great initiative; I call for it in the book, I hope it happens at GSK, consistently, and I hope it is copied”.[iii]
Similarly, in response to GSK signing up to AllTrials, where the Cochrane Institute is one of the partners, and committing to post CSRs, Tracy Brown from Sense about Science said: “GSK signing up to the campaign is very important, off course, because they are a large global player in clinical research so they have a lot of potentially useful information to share, but also because they are finding a way to put in place the infrastructure needed to do this. Which makes it realistic for others and sets a new standard”.[iv]
In a BMJ article published March 2013, CEO Andrew Witty explained our commitment to transparency[v] and our global position is also reflected in the recent submission of our views as part of the consultation process with respect to EMA’s ‘Policy 0070 on publication and access to clinical-trial data’.
We regret that you find a personal meeting irrelevant and would like to repeat our invitation. In GSK we find dialogue very valuable and ultimately to the benefit of patients, and thus our dialogue should be continued in a face to face meeting either in London or in Copenhagen.’
The Chalmers-Vaillance editorial justified GSK-style in part by making a case that patient confidentiality was extremely important and that the risks of identification were great. This is a strategy Pharma have deployed from the start of the current Data Access Debate –
1boringoldman has also covered this ground and the apparent growing links between GSK and AllTrials, and how GSK are using their model of transparency to block access to the data.
[see roaches…]
http://1boringoldman.com/index.php/2013/10/29/an-addendum/
Because there is a mismatch between the CSRs and CRFs, the Study 329 RIATers are at a point where they require the raw data – the CRFs. GSK refuses to hand over the CRFs.
What would the now 35-year-old or so children who participated in Study 329 think should happen at this point?
Now Germany is stigmatizing a portion of the population as filthy, disease-causing vermin, and building fences to keep them away from the Good Germans:
https://www.reuters.com/world/europe/mulled-wine-only-vaccinated-some-german-christmas-markets-2021-11-16/?fbclid=IwAR1F0tLoxd94lu5gnk56DbsPVsEXabCOZ7lD-jYOV0jdGWVeKRghZEmAmG8
What could possibly go wrong?
What’s ‘UP’ Doc…
GlaxoSmithKline poaches Pfizer vax leader Dormitzer after hemorrhaging vaccine R&D talent
by Ben Adams |
Nov 30, 2021 9:00am
https://www.fiercebiotech.com/biotech/glaxosmithkline-poaches-pfizer-vax-leader-phil-dormitzer-after-hemorrhaging-vaccine-r-d
That name is Phil Dormitzer, M.D., Ph.D., who joins the British company as its new global head of vaccine R&D and was previously vice president and chief scientific officer for RNA and viral vaccines at rival Pfizer.
In that role, he worked on Pfizer and BioNTech’s now world-famous COVID vaccine Comirnaty as well as on Pfizer’s experimental RSV vaccine and its early RNA work on influenza (which was also being done alongside BioNTech).
Pfizer refuses to share vaccine knowledge as it announces $US36 billion in vaccine revenue
By Patricia Ranald|November 30, 2021
https://www.michaelwest.com.au/pfizer-refuses-to-share-vaccine-knowledge-as-it-announces-us36-billion-in-vaccine-revenue/
Australian Fair Trade and Investment Network (AFTINET), Amnesty International Australia, Médecins Sans Frontières (MSF) Australia, Oxfam Australia, the Public Health Association of Australia (PHAA) and others rallied outside Pfizer offices in Sydney on November 22 to tell Pfizer to stop profiteering from the pandemic, to stop lobbying against the WTO waiver and to share its vaccine knowledge.
@samizdathealth
@DrDavidHealy
19 of 29 “authors” on Pfizer-BioNTech’s key vaccine study are employees; few are clinicians. ICON, a private CRO, ran the study and wrote the @NEJM article. Our tools for evaluating the product are broken.
Given this reality, “mandates” are madness.
https://rxisk.org/there-was-a-young-woman-who-swallowed-a-lie/
ICON has provided clinical trial services to Pfizer over the past 30 years and formed a strategic relationship with the company
https://www.iconplc.com/news-events/press-releases/icon-pfizer-biontech/
Sir Patrick Vallance is the Chief Scientific Adviser in the United Kingdom. Vallance worked for GSK between 2006 and 2018. By the time he left GSK, he was a member of the board and the corporate executive team. The fines and so on which I have listed took place while Vallance was working as a senior figure at GSK. The last time I looked Vallance still held a big chunk of shares in GSK.
https://www.vernoncoleman.com/malariavaccine.htm
ICON s…
‘But I sense a growing number of doubters,
https://hitchensblog.mailonsunday.co.uk/2021/12/peter-hitchens-posing-in-one-of-our-few-tanks-liz-truss-is-not-the-same-as-being-an-iron-lady.html
Our moronic response to the Omicron threat
I wonder why hardly anyone has pointed out that the anagram of ‘Omicron’ is ‘Moronic’. It is quite easy to work out, and I suspect that the reason is the sort of hushed reverence everyone has now for everything to do with Covid.
Any sort of mockery or criticism of the authoritarian state we are building on the basis of virus-fighting is now treated as a wicked heresy. Voice such doubts and you will be falsely accused, at top volume, of desiring the deaths of the vulnerable and of wanting to let the virus rip, or of being a crazed ‘anti-vaxxer’. But there is something idiotic, if not actually moronic, about the response of this country to the new variant. We simply do not know enough to justify this level of alarm.
As even the pro-panic semi-official newspaper The Times explained on Friday:
‘There is so little data that everything is informed conjecture – back of the envelope calculations with uncertainties so large that the same figures can be used to give succour or presage doom.’
You should know that when The Times says ‘informed conjecture’, it is trying to avoid saying ‘guesswork’.
We have already seen, as I predicted long ago, the ghastly long-term damage done to such things as cancer treatment by the subjection of the whole state machine to Covid priorities.
I mourn for the businesses, small and large, struggling to stay above water through repeated restrictions, and for the children and students whose education has been ripped apart by the same thing.
Not to mention the sinking of this country and Europe into unbelievable and increasingly permanent restrictions on our personal freedom.
I must ask again, have we really got this in proportion? I know many people were, to begin with, completely overcome by the fear of the unknown. But I sense a growing number of doubters, no longer persuaded that the measures work or that they are worth it. Or both.
Make your voices heard, reasonably and patiently, please. It may not save Christmas but it might save Easter.
‘Wall of secrecy’ in Pfizer contracts as company accused of profiteering
US company faces scrutiny over Covid profits after UK agrees to secrecy clause
https://www.theguardian.com/uk-news/2021/dec/05/wall-of-secrecy-in-pfizer-contracts-as-company-accused-of-profiteering
The revelation comes as Pfizer is accused by a former senior US health official of “war profiteering’’ during the pandemic. In a Channel 4 Dispatches investigation to be broadcast this week, Tom Frieden, who was director of the US Centers for Disease Control and Prevention under Barack Obama, said: “If you’re just focusing on maximising your profits and you’re a vaccine manufacturer … you are war profiteering.”
Vaccine Wars: The Truth About Pfizer is on Channel 4 at 7.30pm on Friday
Recently I saw a reply from the GMC addressing concerns raised by an appearance on GMB, by Dr Ellie. The GMC vigoursly defended her position on anti-depressants. Seemingly referring to current NHS guidelines to reinforce their stance.
This week Dr Alex George, an Ambassador for young people’s MH, appointed by the UK Govt took to Twitter to promote destigmatising psychotropic ‘medication’. Under the hashtag ‘Post your Pills’ He posted a picture of himself holding an anti-anxiety pill. His post had 275,000 likes and literally thousands of others posted images showing how proud they were of their pill taking. Dr Alex was then quoted by the UK’s top MH Charity MIND. There were hundreds of images on Minds website too.
The narrative in the main was these pills keep me well, functioning, they save lives!. The analogy of Insulin and diabetes was being used extensively though no mention of a chemical imbalence. More like if I don’t have these Meds I will get unwell, I will be ill.
No where (In the main these posts were by people under 35) was there any mention of serious side effects, the risk of dependence, the risk in pregnancy or the dangers of tapering/withdrawal.
The hashtag should have been ‘Pop a pill for life’ Because that was what seemed to be being advocated.
Quote I take Sertraline, and am unlikely to ever come off it. It makes me function and have capacity to be a functioning member of society. The stigma has to end.
Malin Anderson (Another former Love Island contestant) posts a picture of herself promoting a pill.. revealing she had been suffering pre-natal depression.
Dr Alex’s words :’I really feel we are starting a movement here. Medication Stigma has gone on for far too long. You have the right to control and look after your health.’
Dr Alex has 2 million followers…
With Vaccines being mandated, what next those with depression, anxiety, being mandated to take their Meds in order to function as workers, parents, members of society…
George hardly represents ‘young’ people He looks well into middle age…and talks like a dinasour spouting what he is told to say by his bosses. What people will do for a job eh
Ellie was on a news prog stating several times that she was not an expert on the subject she was asked to contribute to. Obviously been told to be more careful either by the GMC or her agent. But an action being taken to defend Ellie has been quite successful it seems
Have had no reply myself to the formal concern forwarded to them weeks ago.
Sent them a reminder today and asked who is on the decision making panel. Not that there will be any objective investigation of course.
recovery&renewal
@recover2renew
·
36m
Oh yeah The meaning of #safe and effective seems hopelessly corrupted.
Allen Frances
@AllenFrancesMD
9h
I hate Pharma as much as anyone- but there’s no doubt #COVID19 vaccines are miraculously safe & effective. Refusing vaccines now is both foolishly self destructive & selfishly spreads virus to the vulnerable. twitter.com/RxISK/status/1…
RxISK
@RxISK
Oct 18
Not all vaccine-hesitancy is based on politics, or tall tales on Facebook. Patients who’ve already suffered harm from taking approved Rx drugs “as prescribed” find it hard to believe PhRMA promises: https://rxisk.org/prescribed-harms-and-vaccine-hesitancy/
What’s going on Here?
https://davidhealy.org/whats-going-on-here/
“Your job is to change this so both those not getting care and those not getting justice get a better deal”
‘Miraculously’ safe and effective is somewhat close to ‘Remarkably’ safe and effective, as in the GSK Paroxetine Patter…
https://www.bmj.com/bmj/section-pdf/905402?path=/bmj/351/8025/Feature.full.pdf
after a marketing campaign that characterised Study 329 as demonstrating “REMARKABLE Efficacy and Safety.” …
Re comment above dec 5th.
The response from the GMC has been received 2 days later – . I doubt if anyone expected a ‘striking off’ decison .but at least she has been scrutinised once again and likely to have been ‘advised’ off the record.
GMC – E2-5273BC
Inbox
FPD Decisions
Attachments
3:27 PM (2 hours ago)
to me
7 December 2021
Thank you for your complaint form of 5 November 2021.
We have carefully considered the information you provided, and while we appreciate your reasons for writing to us, we don’t feel that these are issues that would warrant further GMC action being taken. We are sorry if this is not the outcome that you were hoping for.
Our Role
Our role is directly related to the registration of doctors. Our responsibilities are all connected to keeping the Medical Register. We oversee medical education; we give entry to the Register for those suitably qualified; we advise on good medical practice while registered; and we remove or restrict registration in response to fitness to practise concerns where there may be a risk to patient safety.
An investigation can only be opened if the concerns raised are so serious that the doctor’s fitness to practise medicine is called into question to such an extent that action may be required to stop or restrict the way in which they can work to protect future patient safety.
The purpose of an investigation is to determine if or to what extent we need to restrict the doctor from working. We are not a general complaints body and we have no legal powers to intervene in or resolve matters for patients.
Our decision
We have been unable to identify any issues that would cause the GMC to launch a full investigation of Dr Cannon’s overall fitness to practise as a doctor.
In reaching this decision we have also taken into account comments a senior, medically qualified member of the GMC staff has made in respect of the information about anti-depressants, as conveyed by Dr Cannon during the ITV ‘This Morning’ television programme.
The Daily Mail article essentially reiterates points Dr Cannon had made about anti-depressants during the ‘This Morning’ programme.
The TV excerpt in question was not a documentary dedicated to establishing the relative pros and cons of anti-depressants. Rather, it was a six-minute piece within a daily ‘magazine-type’ programme that appeared to be designed to provide some ‘general’ information about anti-depressants, and possibly de-stigmatise the use of this medication.
The piece included reference to Dr Cannon’s personal experience of taking anti-depressants.
Dr Cannon mentioned that treatment options (other than anti-depressants) were also available. Most importantly, Dr Cannon advised viewers to speak to their own doctor/GP should they want more information about:
Whether they might be a suitable candidate for taking anti-depressants
The side-effects of taking anti-depressants
Steps patients should take when seeking to come-off anti-depressants
Overall, it would be for a patient’s own doctor/GP to provide advice that was pertinent to the individual patient’s symptoms and circumstances.
We note you have raised your concerns with the programme-makers and with Dr Cannon’s GP surgery (the latter via a local Councillor and the Patient Advice and Liaison Service (PALS) affiliated with a London-based Clinical Commissioning Group) and we consider this to be an appropriate course of action to take under these circumstances.
Reviewing Our Decision
We have an internal review mechanism (‘Rule 12’) which lets us look at certain decisions again. Attached is an FAQ document which explains more about the process
We will only ever review a case if there is a serious mistake with the decision which, if corrected, could lead to a different outcome. Or if there is significant new information that we haven’t seen before, which might have led to a different decision. We would then only review if at least one of those grounds was met and a review is necessary to protect the wider public. Any review needs to start within two years of the original decision, unless there are exceptional circumstances.
To request a review, please complete the attached questionnaire and return either via post or email, using the address provided on the form.
We can only take action when we believe a doctor is not fit to practise and we do not consider this to be the case. We are sorry that we are unable to assist you any further with these matters.
Yours sincerely
The Enquiries Team
On behalf of the Assistant Registrar
The General Medical Council
3 Hardman Street, Manchester, M3 3AW
‘Exotic beliefs’ …
Hardcore vaccine refuseniks could need deradicalising like terrorists – expert
A “deradicalisation” programme like those given to former terrorists or cult members might be the only way to dissuade some hardcore antivaxxers from their beliefs, according to one psychology expert.
https://www.standard.co.uk/news/uk/people-university-of-bristol-hillary-clinton-facebook-newcastle-b970587.html
Around six million eligible people in the UK have not received a single jab a year on from the first Covid vaccination being given in the UK.
Professor Stephan Lewandowsky, chair in cognitive psychology at the University of Bristol, was keen to stress that many of those are likely to be “marginalised” people who are “very difficult to reach for anything”, and for whom access is likely to be the biggest issue rather than psychological or ideological factors.
But for a small section of “hardcore refuseniks”, he said, it may be difficult to reach them by conventional means because their beliefs are so ingrained.
He told the PA news agency: “They’ll refuse anything – ‘I’m not going to wear a mask’, ‘I’m not going to get vaccinated’, ‘I don’t think climate change is happening’, ‘Covid is a hoax’, and, you know, ‘Hillary Clinton is actually a reptilian shapeshifter’.
“You’re getting to people who hold a cluster of very exotic beliefs – now, they’re very difficult to reach.”
Measures that could be used to reach more moderate people who remain unvaccinated, he said, include assigning vaccine appointments to people rather than asking them to book their own, or setting up pop-up clinics in places like supermarkets or shopping centres.
Another measure would be introducing a vaccine mandate, which he said “will disgruntle a few people” but “can be effective”.
But for those with more extreme views, he said, those methods may not be effective.
He added: “In the ideal world, time and money permitting, you can engage even those people in a very slow, long-term process where you affirm their right to have those beliefs… rather than telling them something about themselves they don’t want to hear, let’s put it that way.
“So you tell them something positive, and then engage in what is effectively the same as a deradicalisation process for former terrorists, or cult members.
“Because we’re really talking, when we get down to that small number of committed refusers, we’re talking about the psychology of cults and extremism, and it’s a very similar psychology.”
Mr Lewandowsky said online misinformation is a contributing factor, adding that while some steps had been taken to remove false claims, sites like Facebook and YouTube are “not doing enough”.
“One of the reasons these hardcore refuseniks exist is because they can live in their own ecosystem of misinformation,” he said.
Among those who continue to refuse the Covid vaccines is 44-year-old Paul Barrett from Newcastle who believes they are “dangerous”.
Mr Barrett said he has researched the topic by watching videos and reading articles online and believes the public is “being lied to”.
“Nothing could make me decide to have this jab, it’s dangerous,” he told PA.
“I’ve had Covid and it’s true I was very ill and could barely breathe for over a week but I survived through my immune system, I’m willing to continue to trust said immune system rather than a jab with so many cases of bad, life-ending side affects.
“I believe we’re being lied to.”
Online Covid misinformation is “a very serious public health issue”, according to one expert.
Imran Ahmed, chief executive of the Centre for Countering Digital Hate, told PA: “People have died, and more still are at greater risk of suffering from Covid, because they got their medical information from Facebook and other social media sites.
“These platforms are chronically polluted because anti-vaxxers have been allowed to dump toxic misinformation into people’s feeds on a daily basis for years with impunity.
“Most people who haven’t been jabbed aren’t what you might refer to as ‘committed anti-vaxxers’ – they are merely vaccine-hesitant, because they’ve been deliberately and cynically targeted with a steady campaign of half-truths, baseless conspiracy theories and outright lies.”
Many people who decline being vaccinated research information especially from thebmj which has been updating readers all through the pandemic. Does 44 year old Barrett read it?
On Grounds of Breach of Human Rights
NewsCovid-19: High Court overturns decision to ban GP from posting views on pandemic on social media
BMJ 2021; 375 doi: https://doi.org/10.1136/bmj.n3033
– there seems to be a lot of troubled personalities like Amrin and Stephan who are jumping on a bandwagon , ramping up hate against people they don’t like or who don’t share their views. Pots calling the kettles black – They are behaving like sad rather weird people who sit in their bedrooms dreaming up hate campaigns . Using words lile ‘pollution’ toxic’ ‘terrorists’ against those they diagree with isn’t exactly sophisticated Communications like this from a psychologist is a bit dodgy- shouldn’t Stephan and Amrin be forced to enter a re education programme – not run by Bristol uni though if this is what students are being subjected to.
1 of 2
Reminder: Register to watch the Commission on Human Medicines (CHM) consideration of the isotretinoin review
Inbox
MHRA webmaster@subscriptions.mhra.gov.uk via service.govdelivery.com
10:04 AM (1 hour ago)
to me
CHM consideration of the Isotretinoin Review
View as a webpage / Share
Medicines and Healthcare products Regulatory Agency bulletin
Dear Colleague,
Thank you to everyone who has registered to watch the Commission on Human Medicines (CHM) consideration of the isotretinoin review. We would like to remind those who have yet to register that registration closes on Monday, 13 December 2021 at 17.00 (GMT).
As you may remember, the Isotretinoin Expert Working Group (IEWG) has been reviewing the risk of psychiatric and sexual side effects with isotretinoin. This information is now going to be presented to the Commission on Human Medicines (CHM) where the next step of the isotretinoin review will be considered.
Patients and other stakeholders have been involved throughout the isotretinoin review and we want to continue to involve patients and stakeholders in the next steps.
Although the content and proceeding of CHM meetings are strictly confidential, we are offering patients and other stakeholders who have contributed or have an interest in the review an opportunity to watch CHM’s consideration of the information associated with the review.
The CHM meeting will be held in two sessions. The first session will consider the information reviewed by the IEWG and the second session will consider the IEWG’s recommendations and the regulatory action needed. Patients and other stakeholders are invited to watch the first session which will be recorded and published as part of the review outcome on GOV.UK.
Please note that this CHM meeting is dedicated to the isotretinoin review, it is not a public meeting and only those who have registered will be able to attend.
Date: Friday, 17 December 2021
Time: 14:00-15:30 (GMT)
Register now to confirm your place
Finally, please note that the CHM members must be independent, and we ask that individuals or campaign groups do not directly contact them regarding this review.
We hope you will continue to be involved with this review and you’re able to join the CHM meeting, so please remember to register.
If you have any further questions, please email us at MHRACustomerServices@mhra.gov.uk
Kind regards,
Patient, Public and Stakeholder Engagement Team The Medicines and Healthcare products Regulatory Agency